
We are living in uncertain times. A transitional period, between the ‘antibiotic golden age’, and the oft-predicted ‘post-antibiotic era’; a period where, despite the slow, progressive loss of antibiotic effectiveness, modern life and medicine continues largely unaffected, for now. We are also living in the age of an effective explosion in the capability of DNA analysis. As a scientist in genomics, these are exciting times. They are also deeply worrying ones. Whilst the ability to read and decode the DNA of bacterial species is enabling us to improve the treatment of infections, it is also revealing to us how quickly bacteria are responding and adapting to the antibiotics we are using. The use of this incredibly powerful technique has generated unprecedented insights into the challenges we face as we try to slow antibiotic decline.
The basic techniques to study the response to antibiotics have remained largely unchanged since the time of Fleming. We grow bacteria to test their survival in the presence of different antibiotics. Understanding which genetic elements contribute to resistance has traditionally relied on complex, time consuming sets of experiments, requiring the scientist to identify a potential culprit, remove or inactivate a gene, and re-test the bacteria. Such studies have allowed us to generate public databases of culprit genes, which can be searched with ease. Coupled with Whole Genome Sequencing (WGS), this process can be greatly accelerated.
The first complete bacterial genome published in 19951 heralded a revolution in microbiology. Limited DNA analysis techniques had been around for a while, enabling us to identify the presence of certain, short, DNA sequences. The technique of Whole Genome Sequencing (or WGS) now allows us to read the entire genome of a bacterium. This can be stored and used to analyse the key components - which genes they carry, their similarity to other known strains, and how they are mutating and evolving.
Decoding the first genome took about a year. Today it is possible to sequence 20 bacterial genomes in a day; there are now over 317 million genomic sequences available on the GenBank2 database. In recent years WGS has made the jump from a research-only technology, to one increasingly used in public health, particularly for the surveillance of food-borne pathogens.
A revolution in sequencing-by-synthesis
A significant advance in the transformation of WGS from a research to a public health tool was the development of accessible laboratory protocols and desktop sized sequencing machines, such as the Illumina MiSeq. This uses a technique called ‘sequencing-by-synthesis’ which takes advantage of the fact that DNA is composed of 4 units or ‘bases’ (adenine, thymine, guanine and cystosine – A, T, G, C) which form complimentary pairs (A-T, C-G) – the steps in the DNA ladder.
DNA is extracted from samples using the favoured method of each laboratory, and fragmented into chunks less than 1000 base pairs long. A series of short adaptors and unique barcodes are then ligated (attached) onto each DNA fragment, generating a ‘sequencing library’. The unique barcodes allow different samples to be pooled for analysis, thus saving time without mixing up sample identity. The double-stranded DNA is denatured (separated) into single-strands and loaded onto the sequencing machine. The adaptors allow the DNA strands to attach to the surface of the ‘flow-cell’ inside the machine, which is where the sequencing reaction occurs. An amplification reaction replicates each strand to create a ‘cluster’ of single strands, which act as templates.
After clustering, a solution containing individual bases (A, T, G, C) is sequentially washed over the flow-cell. Bases bind to the single-stranded DNA if the first available base on the template is their compliment pair. In this way, gradually, a complimentary strand of DNA is built for each template. Each time a base binds successfully, the camera equipment within the sequencing machine detects a coloured flash – a different colour for each base. By analysing the order and colour of the flashes, the sequence of the DNA templates – the initial fragments – is decoded, and a whole genome can be reconstructed.
Predicting antibiotic success
Many organisations now have databases of many thousands of bacterial DNA sequences, and their response to antibiotics. These can be analysed to find new DNA mutations associated with antibiotic resistance. Combine this with databases created from knowledge of previous work, and pretty soon you can have enough information to be able to predict which antibiotics will work, based on the DNA analysis alone3, 4, 5.
Is this useful? For bacteria like Mycobacterium tuberculosis, which causes TB (and which can take months to grow and test), genome sequencing has already been shown to be faster and cheaper than standard testing6. Mycobacterium tuberculosis is an important human pathogen, estimated to have infected 9.6 million new people in 2015, and to have killed 1.5 million people. Public Health England plan to adopt WGS for their TB samples; for other, faster growing bacteria, further improvements are required before it becomes viable. However, given current and foreseeable technology it is reasonable to consider that a good portion of our microbiology diagnostics can be replaced by genome sequencing in the next 5 to 10 years.
This technique can also turn up some surprising findings. When large, international TB databases were studied, many TB samples were found to have 'hidden' resistance, not to the commonly-used antibiotics which the labs were testing for, but to second and third-line treatments that weren’t being tested6. The degree of resistance to these 'back-up' antibiotics was not widely known before the study, simply because many labs didn’t routinely look for it- each test adds complexity, time and costs. Determining that back-up antibiotics are ineffective means doctors have to make sure patients get the appropriate treatment from the get-go, and take it religiously, knowing that if first line treatment fails, the 2nd and 3rd line may not be there to help.
Furthermore, stored DNA sequences effectively form a historical archive of the bacteria present at a particular place and time, ready to be searched as new information because available. The power of large database reanalysis was recently elegantly demonstrated with the discovery of the mcr-1 gene, reported in bacteria from animal food sources in China on November 18th 20157. This gene confers resistance to colistin, often used as an absolute last-resort antibiotic, causing concerns that it could combine with many other drug resistance genes to produce a completely untreatable bacterium. Analysis of existing sequence databases subsequently found the gene in samples not only from human guts, but also from, other countries and continents8, 9.
Efforts are ongoing to establish international databases of genome sequences to allow regular surveillance of how resistance is developing and spreading. However there are often conflicting interests. Academic pressures to keep information private until publication, or to avoid being 'gazumped', can limit the sharing of genomic databases, and there needs to be careful curation of quality. However “there can be no real-time surveillance without real-time data sharing”, quotes Frank Aarestrup, one of the leaders in the field. Addressing these issues will be of key importance in maximising the benefits of WGS across the world.
Bacterial ‘detective work’
In 2007 healthcare related infections of Clostridium difficile were at a high, with over 55,000 cases reported in 2007/08 when mandatory surveillance was introduced by Public Health England10. The surge in C. difficile infections that led to mandatory surveillance was widely reported in the UK’s media, which, along with wide reporting of other hospital ‘superbugs’ such as methicillin-resistant Staphylococcus aureus (MRSA) led to accusations of poor staff hygiene and dirty hospitals facilitating the spread of healthcare associated infections.
In the case of C. difficile, DNA analysis showed the story was not quite as simple. When the DNA sequences of the C. difficile bacteria from hospital patients were compared, it suggested that only around a third were actually genetically related (suggesting there had been transmission from patient to patient). In reality there were so many genetically diverse strains, it suggested that there was a whole lot of C. difficile out in wider world, living, evolving independently, and probably never coming near a hospital11. Finding superbugs, it seems, required one to cast the net far wider.
Other times, instead of ruling transmission out, WGS could be used to definitively confirm it. Traditionally, understanding how M. tuberculosis spreads has been left to a combination of ‘detective work’ by specialists tracking where a patient has been and when, and limited tests which identify similar bacteria. When four English hospitals sequenced the DNA from their mycobacterial cultures, they found cases where there were no identified links between patients, but the DNA from bacteria were so similar that it was almost certain the patients caught the infection from the same person6, 12. This sort of information means that infection specialists can go back and investigate in detail, find the ‘cryptic’ source, and ensure they are treated to prevent further spread.
Using similar techniques, researchers have been able to track ‘superbug’ outbreaks not just to patients or healthcare workers in the case of MRSA13, but even to dogs14, and Pseudomonas aeruginosa infections in burns units to water sources15. The power of the 'bacterial fingerprint' can be of great assistance in tracking down sources of infection, and stopping outbreaks. Importantly, these analyses can be done using the same DNA sequences used to study antibiotic resistance - after the hard work of DNA sequencing, additional tests are effectively 'free' as they are discovered and refined, limited only by the computing power required to interrogate the stored information.
How is resistance evolving?
One thing that has been difficult to reconcile with our existing understanding of antimicrobial resistance is the speed at which it is developing. In species like M. tuberculosis, we can see how resistance to an antibiotic occurs due to random, but useful, mutations in the genetic code, the 'beneficial mistake'. However in other cases doctors have seen rapid development of resistance to multiple antibiotics, caused by entirely new genes, in totally different strains and species. And this resistance can go from rare to endemic in as little as a few years.
How was this possible? One thing that WGS has helped us understand is just how much 'shuffling' of DNA can go on in, and between, different bacteria. Bacteria can share DNA - transferring it between different individuals on small DNA loops, called plasmids. One plasmid can contain the necessary genes to inactivate multiple different antibiotics - multi-drug resistance in one handy, mobile packet. DNA can even 'jump' between plasmids, or to different locations on the main bacterial genome, on short, incredibly mobile DNA sequences, called transposons.
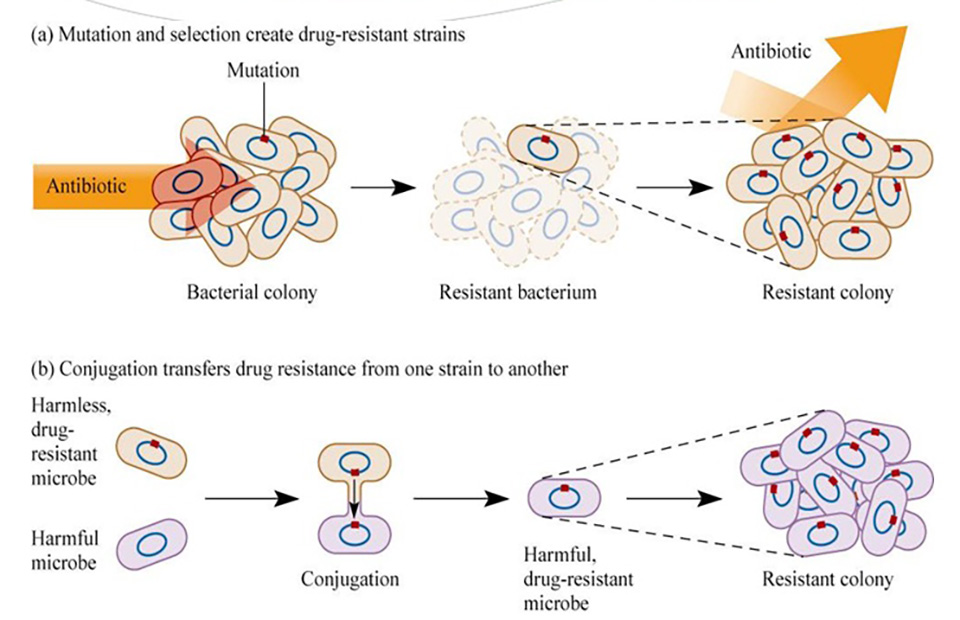
By comparing the DNA of multiple different bacteria from the environment, as well as from patients, it is sometimes possible to reconstruct a 'backstory' of how resistance came to be. OXA-48 is one of the genes that inactivate carbapenems, which are one of our last-line effective antibiotics used to treat multidrug-resistant bacteria. Evidence from bacterial DNA analysis suggests that this gene probably started out in a harmless river bacterium, possibly one of the Shewanella species, and then jumped to an entirely different species on one of these mobile DNA elements. At some point, OXA-48 then managed to team up with a Klebsiella pneumoniae strain particularly good at surviving and spreading in hospitals. K. pneumoniae can cause urinary and bloodstream infections, and this strain is currently giving much of southern Europe a major headache, becoming endemic in the space of a few years16.
We know that antibiotics can enter the water system, from sewage, directly from factories and through run-off from farms. Up to 80% of all antibiotics in the US are being used, not in humans, but in the farm industry17. It is not unreasonable to speculate that mixing antibiotics, environmental and human-associated bacteria is the perfect melting pot to encourage antibiotic resistance genes to spread to new species. There is highly suggestive evidence that this mobile DNA can also be shared between bacteria infecting farm animals and bacteria infecting humans18. Transmission may well be happening in both directions. Resistance is developing and spreading, it appears, not just in humans, but in many ecosystems where bacteria are combined, often unthinkingly, with antibiotics.
Can WGS help to slow antibiotic decline?
The future of WGS itself is a bright one. The technology is getting faster and cheaper by the day, and it has begun to transition from a research technique to one that is replacing traditional microbiology. From this point of view the next step is to speed up the process to the point where it can provide real-time, or same-day analytics. New developments, such as the Oxford Nanopore (TM) technology, suggest this is viable within the next 5-10 years. It is indeed highly likely that WGS will make large steps in improving the diagnosis and treatment of patients with infections in the next few years. Steps are being taken to integrate, and share, genomic information across borders, to help track the spread of resistance internationally.
Whether the lessons of WGS will be able to slow antibiotic decline remains an open question. Response is required at a national, governmental level, to respond to the careless, and often unnecessary pollution of our biosphere with antibiotics. Reducing use in humans, animals, and controlling release into the environment are priorities. Without action, it is scarce comfort that we will be able to observe and understand the loss of antibiotics in intricate detail.
It is hard not to feel a sense of awe of the ingenuity of the bacterial machine in surviving human intervention, as you see it revealed in DNA sequencing; findings that are beautiful in their science, and often deeply concerning in their potential consequences. For the world of bacterial genomics, these are indeed, exciting, uncertain times.








